Compared to active-site-binders, allosteric inhibitors have the advantage of being active against resistant enzymes with active-site mutations and may have increased specificity for a given sub-family of enzymes. To search for allosteric drug-target sites, we study the structural basis of enzyme activation in specific cellular processes. Our strategy is focused towards identifying conformations of the enzyme ― present either stably or transiently during activation ― that are specific to a given biological pathway. These conformations can be targeted to develop pathway-specific inhibitors.
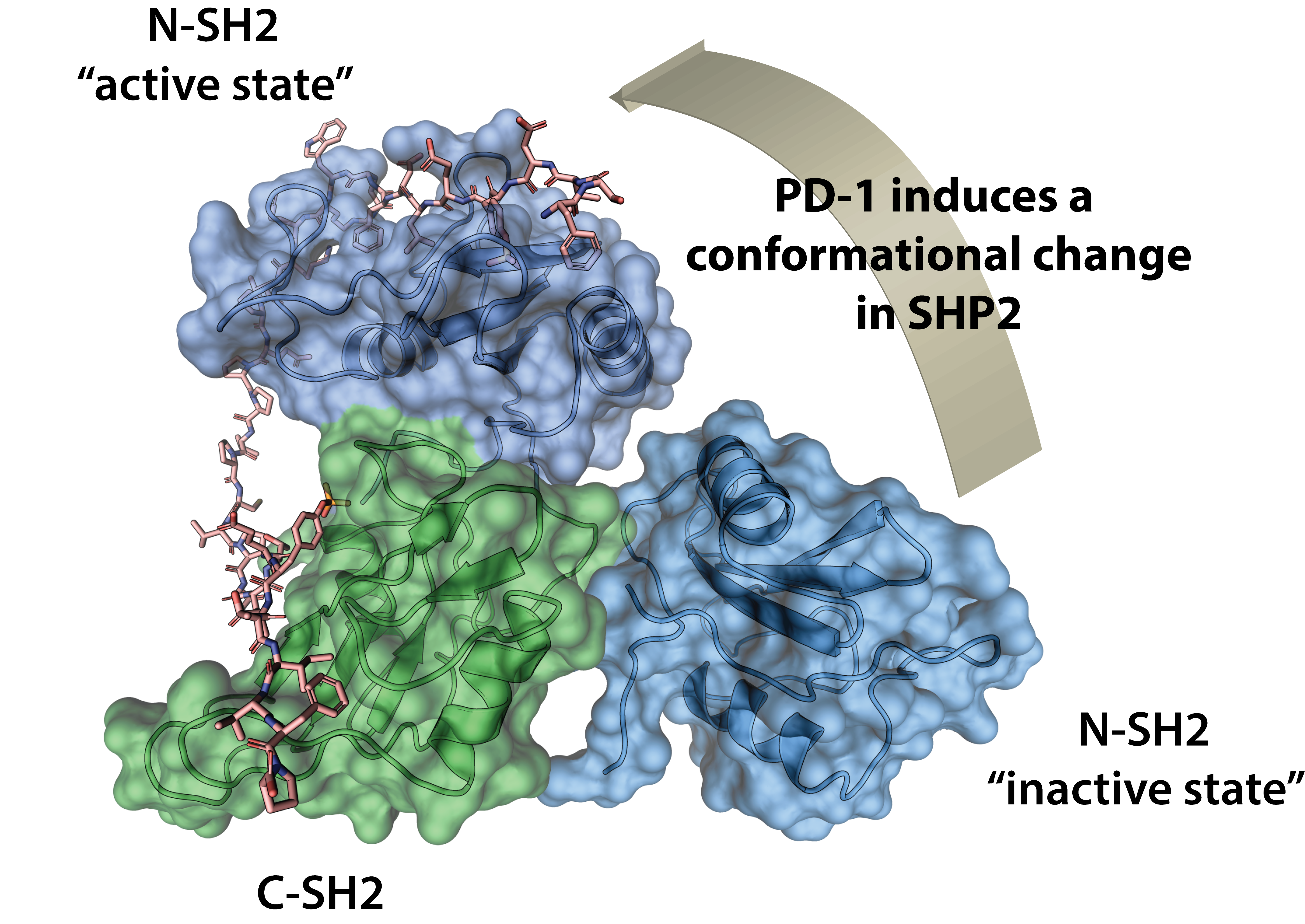
As an example of these efforts, we have studied the PD-1-dependent activation of SHP2 during the process of cancer immune escape. The cytoplasmic Src-homology-2 (SH2) domain-containing protein tyrosine phosphatase 2 (SHP2) is involved in various kinds of leukaemia and solid tumours and is also a key mediator of inhibitory receptor signalling through its interaction with immune checkpoint receptors such as PD-1. The success of anti-cancer therapy based on monoclonal antibodies that disrupt PD-1 signaling suggests that blocking the SHP2–PD-1 interaction by targeting SHP2 may result in an efficient and inexpensive anti-cancer strategy. This hypothesis is supported by many reports demonstrating the role of SHP2 in T-cell activation.
Using a combination of structural and computational methods, we have established the structural details of the PD-1-mediated SHP2 activation and identified three routes to modulating SHP2 activation (Marasco et al., Science Advances, 2020; see figure). Currently, we are focusing on one of the SHP2 structural models determined during this work to develop small molecules that are capable of down-regulating PD-1-dependent SHP2 activation.