Small molecules can regulate the function of proteins, nucleic acids or even complex molecular machines. The development of specific binders that selectively alter the function of only one or a few cellular targets relies on the availability of structural information. When this structural information is not available, the rational design of selective drugs is heavily impeded. In this respect, drug-discovery is often limited by the lack of structures of the target receptor in complex with the lower-affinity ligands (lead compounds) that are identified by high-throughput screening or by fragment-based lead discovery.
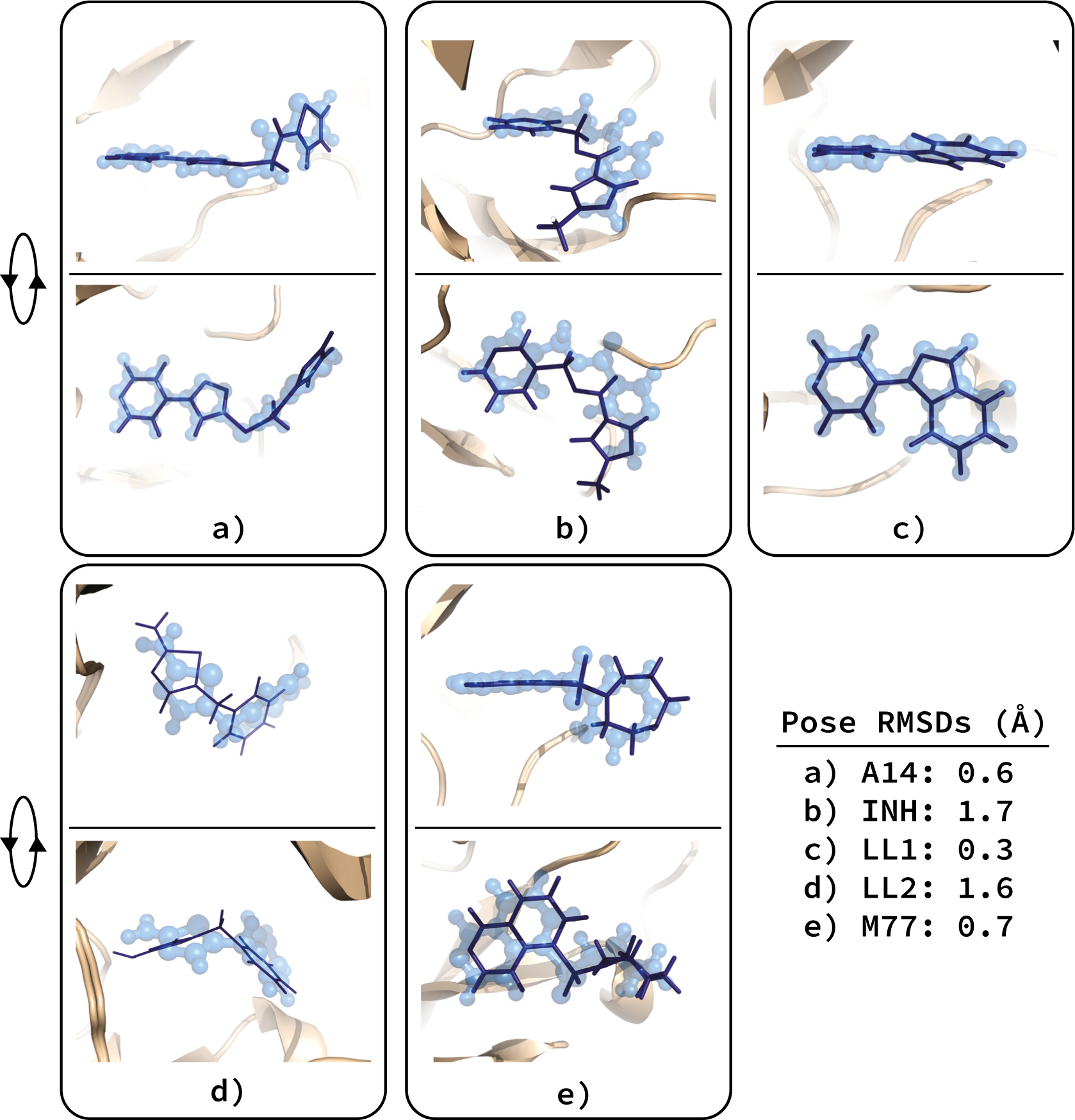
INPHARMA, an NMR-based methodology that we developed a few years ago, alleviates this problem. INPHARMA (Interligand NOEs for PHArmacophore Mapping) provides access to the relative binding modes of low-affinity ligands to a common target when a structural model of the receptor in its apo state is available. The method is based on the observation of inter-ligand, doubly-transferred NOEs (nuclear Overhauser effects) between two competitively binding ligands L1 and L2. Such NOEs do not originate from direct transfer of magnetization between L1 and L2, but rather from a spin-diffusion process mediated by the protons of the receptor binding pocket and are therefore dependent on the specific interactions of each of the two ligands with the protein. The experimental information derived from the INPHARMA NOEs is used to select the correct binding poses from a set of many possible binding orientations generated by molecular docking (see figure).
INPHARMA has been validated on a system consisting of multiple ligands binding the catalytic subunit of the Protein Kinase A (PKA) and of Cyclin-dependent Kinase 2 (CDK2) in collaboration with Sanofi-Aventis (Orts et al., Angewandte Chemie, 2008; Skjærven et al., J. Am. Chem. Soc., 2013; Sikorska et al., MedChemComm, 2015) and has been used to determine the binding modes of epothilone A, discodermolide and tubulysin to tubulin (Reese et al., Angewandte Chemie, 2007; Sánchez-Pedregal et al., Angewandte Chemie, 2006; Kubíček et al., Angewandte Chemie, 2010).